Jake Kim, Moses Lee and Cathy Lee Park Systems Corp., Suwon, Korea
Translated by Cherie Jung, Chie Goto
概要
細胞サンプルの保存プロセスにおいて、細胞の固定は幅広い生物学的アッセイに不可欠な要素の一つです。このプロセスの重要性を判断するために、原子間力顕微鏡(AFM)や走査型イオンコンダクタンス顕微鏡(SICM)などの走査型プローブ顕微鏡(SPM)技術を使用して、生細胞と固定細胞の比較分析を行い、機械特性と表面変動の測定をそれぞれ行いました。これらの技術を駆使して、細胞固定が細胞の機械特性にどのように影響するかを幅広く理解し、 細胞固定プロトコルを確立するための新しい機会を得ました。
はじめに
インビトロ研究のための細胞固定の主な目的の一つは、細胞の本質的な化学的および物理的特性を維持 することにより、細胞または細胞成分を本物そっくりの状態で保存することです。細胞固定は、抗体が細胞内構造にアクセスできるようにすることで免疫染色にも役立ちます[1]。生細胞と固定細胞を比較すると、固定細胞は細胞表面全体でより均一な構造を維持していることがわかります[2]。しかし、生細胞と固定細胞の二つの機械特性変化の間には、系統的な 評価や相関関係はありません。細胞固定プロトコルのさまざまな状態を観察することにより、細胞固定のプロセスを行う上で重要な知識を得ることができます。細胞膜と細胞質タンパク を架橋 するために多くの固定剤が利用可能であり、パラホルムアルデヒド(PFA)が細胞や組織に 最も広く使われています。PFAは、分子を共有結合で架橋 し、それらを結合して、不溶性の網目構造を作ることによって、細胞表面の機械特性を変化させます。 PFAで固定された生細胞と固定細胞 の機械特性を調べるために、原子間力顕微鏡(AFM)と走査型イオンコンダクタンス顕微鏡(SICM)を使用して、それぞれ弾性率と表面変動を測定しました[3-6]。
実験のセットアップ
細胞サンプル
サンプルは、マウス線維芽細胞L929(ATCC、USA)を10%ウシ胎児血清(Thermo Fisher Scientific、US)と1%ペニシリン/ ストレプトマイシン(Invitrogen Life Technique、USA)を加えた(DMEM; Dulbecco’s modified eagle medium, Invitrogen Life Technique、USA)培養液中、37°C、加湿雰囲気、5%CO2濃度で培養したものを用いました。 細胞(密度 1 x 104/mL)を直径35mmの細胞培養ペトリ皿(NUNC、デンマーク)に取り 、リン酸バッファー (PBS、Sigma-Aldrich、米国)で3回リンス した後、4%PFA溶液で5分間処理しました。AFMおよびSICM実験を行う前に、固定細胞 をPBSで3回リンスしました。
AFMとSICMのセットアップ
細胞イメージングのセットアップは、生物学的用途向けに特別に設計された倒立顕微鏡 (Nikon Corp.、日本)を備えたPark Systems走査型プローブ顕微鏡(Park NX-Bio、Park Systems、韓国)で構成されます。AFMは、SICMを使用し、サンプルの機械特性と柔らかい サンプルの細胞表面情報を収集しました。すべての実験は、 37℃、5%のCO2、95%湿度に調整された チャンバー内 で、生細胞培養を維持するために必要な環境で行われました。
細胞のヤング率のAFM測定
細胞のヤング率を推定するためAFMでフォースカーブを取得します。 バネ定数の公称値が0.09N/m 程度の市販のカンチレバー(BL AC40TS、オリンパス、日本)を使用しました。バネ定数が小さいカンチレバーを使うと、小さな力を加えるだけで比較的大きなカンチレバーの変位を取得することができ、 細胞表面に対し、信頼性の高い データを収集することができます。AFMカンチレバーのバネ定数の較正 は、熱振動法にて行われ、それぞれ512のデータポイントを含む50のフォースカーブを使って行われました(図1a)。フォースカーブを分析するために、Park Systems解析ソフトウェア(Park XEI、Park Systems)内のHertzモデル を用いました。AFMチップの形状は、1/2開き角 αの4面ピラミッドが想定され、その結果、カンチレバーにかかる力(F)は次のように表すことができます:
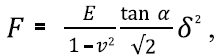
ここで、Eはヤング率、νはポアソン比、δは押し込み深さ を表しています。ポアソン比は0.5に設定され、αは35℃に設定されました。AFMスキャンレートは1µm/sに設定され、最大加重 は8nNでした。
細胞イメージングおよび変動分析のためのSICM測定
SICMは、ナノピペット内に入れた電極と液中にある電極 の間を流れるイオン電流を利用します(図1b)。イオン電流は、チップからサンプルまでの一定の距離を調整し、ナノピペットがトポグラフィー情報をスキャンするための フィードバック信号となります 。プローブの特性上、横方向の分解能は高くできませんが(~30nm)[7]、 SICMは、サンプル表面に機械的な力を加えることなく、有益な トポグラフィー測定値を示すことができます 。図2は、L929の様細胞と固定細胞の形状と実際の高さを表した画像です。 一見、二つのケースに違いはみられませんが、固定細胞の表面は、細胞膜タンパク質の架橋結合のためにわずかに粗いことが わかります。SICMイメージングとイオンカレントディスタンス(ID)カーブの実験を行うために、CO 2 レーザーピペットプラー(Sutter Instruments、USA)を用いてホウケイ酸キャピラリー(内径0.6mm、外径1.0mm、World Precision Instruments, SSA)から内径80nmのナノピペットを製造しました。
結果と考察
生細胞と固定細胞のヤング率
生細胞と固定細胞表面の剛性を調べるために、生細胞、固定細胞とリファレンスとしてディッシュの面について、フォーススペクトロスコピーの測定値を取得しました。 これらのフォースディスタンスカーブは図3aに示されています。固定された細胞は、生細胞と比較して著しく急峻な フォースカーブを描いています。さらに、表面の押し込みに必要な力は、生細胞よりも固定細胞の方が大きいことが わかります。表1では、固定細胞の剛性(77.95 kPa)が生細胞(8.11 kPa)よりも大きいことを示す平均ヤング率の値を表しています。これらのAFMによる細胞の剛性測定は、繊維状の構造が 剛性に強く影響している ことを示して います。もう一つの注目すべき点は、PFA処理がF-アクチンフィラメントを含め細胞表面タンパク質の架橋 に影響を与えることです。さらに、細胞の硬化はタンパク質の架橋 に対応することも注目すべきポイントです[8]。さらに言えば、PFA固定プロセスは、細胞表面で利用可能なランダムに分布した架橋 部位の数に応じて、細胞剛性の増加に直接関連しているとみなすことができます[9]。
生細胞と固定細胞の表面変動
細胞表面の変動を測定するために 、生細胞、固定細胞、および基板(ディッシュ) でのSICM測定を行うことで I-Dカーブを取得し ました。I-Dカーブは、基板(ディッシュ) で最も急な勾配を示していますが、未処理の場合にはより 幅の広い勾配をみせます。PFA処理された細胞の場合、I-Dカーブはこれら二つの中間の勾配を示します 。その後、生細胞は 固定細胞と比べてより多くの活性を示した と結論づけることができました。PFA処理プロセスにより、固定細胞の表面変動が小さくなり、細胞膜と細胞質タンパク の間でタンパク質が架橋されます 。
まとめ
基本的な機械特性の 違いは、生細胞とPFAで固定された細胞を比較することによって実証され ます。原子間力顕微鏡および走査型イオンコンダクタンス顕微鏡測定により、PFAで処理した 際の細胞の表面変動および弾性率の明確な遷移が明らかになりました。PFAで完全に固定すると、生細胞と比べて細胞表面の変動が減少し、ヤング率は5倍に増加しました。これらの調査結果 により、細胞がPFAによる化学処理にどのように反応するかについての重要な 知見を得ることができました。細胞に対するPFAの化学的影響に関する基本的な知識に加えて、この実験では、細胞表面の機械特性に対するPFAの影響が明らかになりました。細胞膜は柔軟で可変であると想定されていますが、特定の状況(特に化学処理)では、生物学的および形態学的レベルでの変化が生じます 。このような観察は、細胞のダイナミクスに関する細胞機能を理解するために必要なもので 、細胞表面の変動をさらに研究するための強い動機も与えます。走査型プローブ顕微鏡技術、特にAFMとSICMは、固定細胞と生細胞の両方の定量的研究の応用において重要なツールになります 。
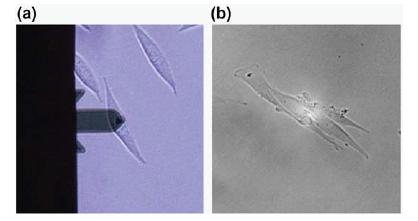
図1: AFM(a)およびSICM(b) のL929セルの光学画像。各プローブは、単一細胞の頂点に配置されています 。
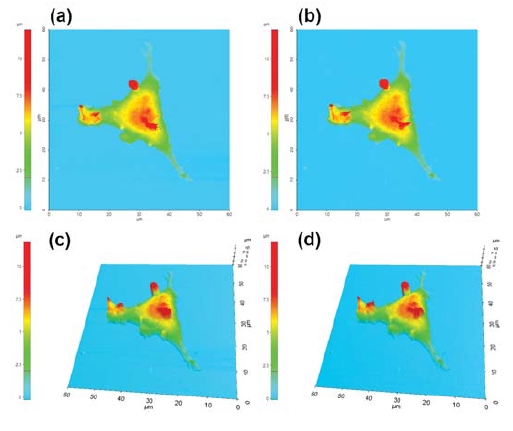
図2: SICMによる生細胞(a)、(c)および4%PFA処理をした細胞(b)、細胞表面イメージング(d)
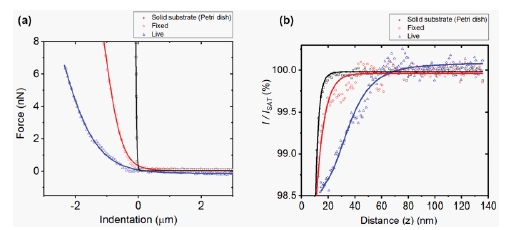
図3: ディッシュ基板(黒)、4%PFA処理した(固定)細胞(赤)と生細胞(青)の平均フォースディスタンスカーブ(a)、イオンカレントディスタンスカーブ(b)
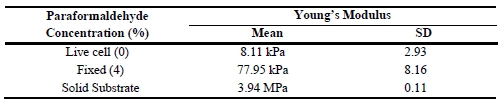
表1:ヤング率の値
参考文献
1. Lanier, L. and N. Warner, Paraformaldehyde fixation of hematopoietic cells for quantitative flow cytometry (FACS) analysis. Journal of immunological methods, 1981. 47(1): p. 25-30.
2. Yamane, Y., et al., Quantitative analyses of topography and elasticity of living and fixed astrocytes. Journal of electron microscopy, 2000. 49(3): p. 463-471.
3. Binnig, G., C.F. Quate, and C. Gerber, Atomic force microscope. Physical review letters, 1986. 56(9): p. 930.
4. Korchev, Y.E., et al., Scanning ion conductance microscopy of living cells. Biophysical journal, 1997. 73(2): p. 653.
5. Cappella, B. and G. Dietler, Force-distance curves by atomic force microscopy. Surface science reports, 1999. 34(1): p. 1-104.
6. Mizutani, Y., et al., Nanoscale fluctuations on epithelial cell surfaces investigated by scanning ion conductance microscopy. Applied Physics Letters, 2013. 102(17): p. 173703.
7. Rheinlaender, J., et al., Comparison of scanning ion conductance microscopy with atomic force microscopy for cell imaging. Langmuir, 2010. 27(2): p. 697-704.
8. Hopwood D. Theoretical and practical aspects of glutaraldehyde fixation. InFixation in histochemistry, Springer, Boston, MA. 1973: p. 47-83.
9. Tanaka KA, et al., Membrane molecules mobile even after chemical fixation. Nature Methods. 2010 Nov;7(11):865.